

TEL:17312606166(魏經理)



美鳳力臨床前大動物實驗中心

 17312606166
17312606166
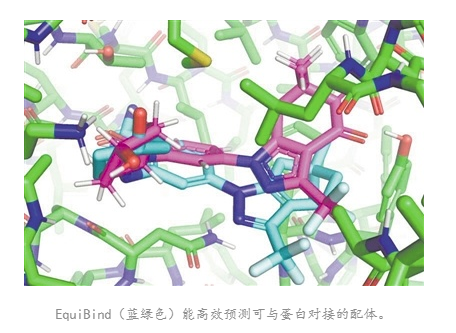
據美國麻省理工學院(MIT)官網月7月12日報道,該校科學家開發出一款名為EquiBind的幾何深度學習模型,其將類藥物分子與蛋白配對的效率比現有最快的計算分子配對模型QuickVina2-W快1200倍。相關研究已經提交預印本服務器,并將提交給國際機器學習大會。
在藥物開發之前,研究人員必須找到有潛力的類藥物分子,這些分子可以與某些蛋白質靶點正確結合或“對接”——這一過程被稱為藥物發現。類藥物分子(配體)成功與蛋白質對接后,可以阻止蛋白質發揮功能。如果蛋白質是細菌的一種必需蛋白質,配體就可以殺死細菌,從而保護人體。
目前尋找潛在藥物候選分子的計算過程大致如下:大多數最先進的計算模型依賴繁重的候選采樣,以及評分、排序和微調等方法,從而讓配體和蛋白質之間實現最佳“匹配”。
最新研究主要作者、美國麻省理工學院電氣工程和計算機科學系研究生漢尼斯·斯塔克表示,上述傳統的配體—蛋白質結合方法就像“嘗試將鑰匙插入有許多鎖孔的鎖中”。這種方法需要花費大量時間對每個“鎖孔”進行嘗試,才能找到最佳匹配。相反,EquiBind僅需一個步驟就可以直接精準預測配體與蛋白質配對的精確位置,這是因為其擁有內置的幾何推理能力,可以幫助模型了解并學習分子的基本情況,在遇到新的數據時能夠進行概括,以做出更好的預測。
該研究引起了專業人士的興趣。接力醫療公司首席數據官帕特·沃爾特斯建議其團隊在現有的一種用于肺癌、白血病和胃腸道腫瘤的藥物和蛋白質上嘗試這一最新模型,結果EquiBind取得了成功,而大多數傳統的配對方法無法讓蛋白和配體成功配對。
沃爾特斯說,EquiBind為配對問題提供了一種獨特的解決方案,它結合了姿態估計和結合位點識別。這種方法利用了數千種公開可用的晶體結構的信息,可能對藥物開發領域產生新的影響。
| 上一篇:細菌混合微型機器人可在體內遞送藥物 未來有望執行抗癌任務 | 下一篇:斑馬魚心臟毒性模型:藥理學評價 |
|
|
|
|
