

TEL:17312606166(魏經理)
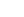



美鳳力臨床前大動物實驗中心

 17312606166
17312606166
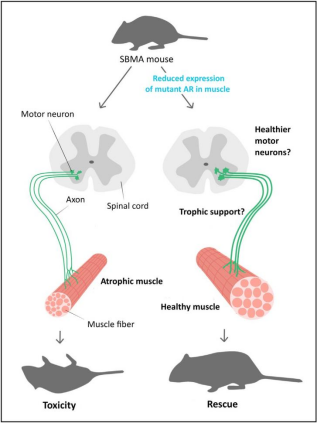
肯尼迪病(Kennedy’s disease, KD)又稱為脊髓延髓肌萎縮癥(Spinal and bulbar muscular atrophy, SBMA),是一種X連鎖、成人發病型運動神經元疾病,其特征是延髓和四肢肌肉的緩慢進行性無力。肯尼迪病主要影響成年男性,估計本病的發病率(1~2)/10萬,相當多的患者可能被誤診為其他神經肌肉疾病,包括肌萎縮側索硬化癥。 世界各地有不同種族背景的SBMA患者報道。一般為30~60歲發病,疾病初期多為非特異性癥狀,如姿勢性震顫和肌肉痙攣。姿勢性震顫多出現在患者肢體無力前數年甚至數十年前,值得引起臨床醫生重視,有研究發現其原因可能與臨床下的感覺障礙和運動單位減少有關。早期臨床癥狀包括面部收縮震顫或言語不清,多有肌束震顫,尤其是口周和舌頭。 雖然目前認為肯尼迪病為下運動神經元病,但也有研究表明,肯尼迪病患者存在輕度認知功能障礙,表現為言語流暢性障礙、概念形成異常以及記憶力下降。部分肯尼迪病患者表現出雄激素不敏感的跡象,如乳房發育、睪丸萎縮、勃起障礙、生育能力降低。女性攜帶者通常無癥狀。體格檢查發現以下運動神經元體征為主,可見輕度的肌肉萎縮、束顫,肌力輕度減退,肢體近端更明顯,腱反射減弱或消失和感覺缺失。大多數患者血清中的肌酸激酶水平升高,部分合并高血壓、高血脂癥、輕度肝功能異常、葡萄糖耐受不良等。肯尼迪病患者電生理檢查以廣泛的慢性神經源性損害為最主要改變,多伴感覺和運動神經傳導異常,且感覺異常比運動異常更多見[1]。 肯尼迪病的臨床特點[2] 肌肉病理檢查 A:患者肌肉活檢HE染色顯示肌纖維呈重度萎縮,萎縮纖維呈束狀分布,間質脂肪增多,未見炎細胞浸潤;B:肌肉活檢NADH染色顯示Ⅰ型纖維成組化現象[3] 有學者為了研究致病蛋白AR的特定配體——睪酮在SBMA中的作用,在巨細胞病毒增強子和雞來源的β-actin啟動子的控制下,獲得了表達含有97個CAGs的全長人AR的轉基因小鼠,該模型不僅再現了神經功能障礙,而且還再現了與性別的表型差異,這是SBMA的一個特殊特征[4]。 又有學者鑒定了兩個跨越AR基因整個長度的重疊的BACs,通過重組策略,將兩個BAC融合,創建了一個AR BAC結構,除了所有8個AR外顯子外,還包括DNA50到第一個AR外顯子的50kb和DNA30的30kb到最后一個AR外顯子,通過這種重組方法,該學者還引入了一個121CAG重復通道,并在AR外顯子1兩側設計了兩個loxP位點,構建了一個浮動的AR CAG121 BAC(BAC fxAR121)轉基因結構體,然后獲得了BAC fxAR121轉基因小鼠,經過表達發現與YAC CAG100(YAC AR100)小鼠的hAR RNA、mAR內源性蛋白水平表達水平相當[5]。有學者通過此模型小鼠研究確定了肌肉是突變AR毒性的一個部位,并建議靶向該組織中的突變蛋白表達作為治療該疾病的一種方法[6]。 減少肌肉中突變體AR的表達在SBMA小鼠模型中具有有益的作用[6] 治療方式 ? 對癥治療 對癥治療有助于緩解震顫、內分泌異常、肌肉痙攣、呼吸衰竭、吞咽困難等癥狀。對已確診為SBMA的患者應進行長期隨診觀察。對于痛性痙攣,鎂劑,替扎尼定、巴氯芬、加巴噴丁、丙戊酸鈉,卡馬西平等均可選用。若患者存在糖尿病,則按照現行診療原則進行治療。若患者因吞咽困難出現營養不良,可行經皮內鏡胃造瘺;對于小部分出現呼吸功能障礙的患者,無創正壓機械通氣可以改善患者癥狀;若患者晚期出現呼吸功能衰竭,必要時可根據患者意愿決定是否行機械輔助通氣[7]。 ? 特異性治療 對于包括肯尼迪病在內的多聚谷氨酰胺疾病,多個機制可能參與神經元功能障礙和最終的細胞死亡。它們包括:錯誤折疊導致疾病的蛋白質功能改變;由突變型蛋白從事有毒性的蛋白質的相互作用;形成有毒的寡聚物;轉錄失調;線粒體功能障礙導致受損的生物能學和氧化應激;受損的軸突運輸;異常的神經信號,包括興奮性毒性;細胞蛋白質穩態受損;RNA毒性。雖然這些分子機制都有可能作為治療靶點,但因為肯尼迪病的致病機制尚未闡明,故目前仍沒有標準的治療方案,現有方案大都只停留在動物模型實驗階段。在這些治療方法中: 1.雄激素剝奪已經應用到臨床試驗。一些動物實驗已表明,雄性肯尼迪病小鼠的組織病理學發現AR蛋白表達水平與血液中睪酮水平呈正相關。為了支持這一觀點,在肯尼迪病的小鼠模型中,手術去勢顯示反向運動功能障礙。類似的結果也在亮丙瑞林的臨床前研究中得到。亮丙瑞林是一種促黃體激素釋放激素衍生物,可抑制垂體釋放促性腺激素,抑制睪丸釋放睪酮。這種藥物已被用于各種性激素依賴性疾病,包括前列腺癌、子宮內膜異位癥。在雄性肯尼迪病小鼠,亮丙瑞林成功抑制了致病的AR核積聚,導致神經肌肉的表型明顯改善。亮丙瑞林的雄激素阻斷作用也由前列腺和精囊的重量減少得到了進一步證實。與對照組小鼠相比,亮丙瑞林治療的肯尼迪病小鼠表現出更長的壽命、較大的尺寸、更好的運動性能。亮丙瑞林的2期臨床試驗顯示,在亮丙瑞林治療大約2~4周后,人血清睪酮水平下降到了手術去勢水平。與安慰劑組比較,這種藥物治療的患者在陰囊皮膚活組織檢查(活檢)顯示突變的AR積累減少,吞咽功能增強。接受亮丙瑞林治療的患者尸檢結果表明,雄激素剝奪抑制了脊髓和腦干運動神經元AR的核積。這些觀察結果表明,亮丙瑞林通過給藥抑制了突變體AR在肯尼迪病的神經肌肉毒性蓄積。 2.AR共同調節因子也作為替代治療目標,因為它們控制細胞AR的功能和分布。從生姜、咖喱中萃取的化合物5-羥基-1,7-雙(3,4-二甲氧基苯基)-1,4,6-heptatrien-3-1(AsC-J9)可擾亂AR和其共同調節因子的相互作用。近期研究發現AsC-J9可選擇性降解AR蛋白并減少其在細胞內積聚,使AR-97O轉基因小鼠的運動能力得到顯著改善,平均生存期由28周增至39周,且AsC-J9治療后的肯尼迪病小鼠,血清睪酮水平基本正常,性功能和生育能力顯著改善。 3.細胞防御機制的激活是肯尼迪病另一個有希望的治療方法。在肯尼迪病的小鼠模型中,Hsp是一種應激誘導的分子伴侶,可分為不同的家族:Hsp100、Hsp90、HspT0、Hsp60、Hsp40和小Hsps。在肯尼迪病的小鼠模型中,Hsp在多聚谷氨酰胺疾病中(包括肯尼迪病)的高水平表達可抑制異常蛋白的毒性積聚,并可通過多種途徑阻止細胞死亡。故通過藥物誘導方法增加其表達水平,很有可能成為治療肯尼迪病和其他多聚谷氨酰胺疾病的一種新方法。替普瑞酮(GA)在多種組織中都可強烈誘導Hsp的表達。予肯尼迪病轉基因小鼠口服GA后可顯著上調HspT0在中樞神經系統中的表達水平,并可抑制致病AR蛋白在細胞核內的積聚,從而顯著改善神經肌肉相關的癥狀表現。另一方面,在肯尼迪病中,抑制Hsp90已被證實通過激活泛素-蛋白酶體系統抑制神經變性。在肯尼迪病的細胞和小鼠模型中,用一種強效的Hsp90抑制劑-17-烯丙基氨基格爾德霉素(17-AG)進行治療,促進了致病AR蛋白酶體的降解。 4.轉錄失調是用于治療干預的另一個靶點。抑制組蛋白去乙酰化酶(HDAC)活性可增加組蛋白乙酰化,隨之增加基因的轉錄水平,HDAC抑制劑已被認為在多聚谷氨酰胺疾病中有治療作用。丁酸鹽是被發現的第一個HDAC抑制劑,在肯尼迪病小鼠模型中,丁酸鈉通過口服給藥,可上調神經組織中組蛋白乙酰化的表達,改善模型的癥狀和病理表型[8]。 賽業生物基因治療一站式解決方案 近年來全球多款基因治療藥物相繼獲批上市,還有多種針對不同適應癥的基因治療藥物正處于臨床研究階段,基因治療已然為眾多亟待拯救的患者帶來新的治療希望! 賽業生物基因治療一站式解決方案可為從事基因治療的研究者提供更高效的基因功能解析與基因治療一站式整體解決方案,包括靶點篩選與功能研究,動物模型構建和病毒載體如AAV、LV、ADV等設計與包裝,以及表型分析等全流程服務。 RDDC助力罕見病研究 罕見病數據庫(以下簡稱“RDDC”)由罕見病基因治療聯盟理事長單位——清華珠三角研究院人工智能創新中心主持開發,并由副理事長單位賽業生物提供生物遺傳技術支持,歷經1.0至2.0版本升級,可為用戶提供相應罕見病的信息,并更好的服務科研人員對于數據查詢和數據挖掘的需求。 *聲明:RDDC數據和工具僅為科研使用,僅供參考,不可作為醫學診斷和評判的最終定論。 
——機理研究")
——機理研究")
參考文獻及引用圖片來源:
[1]Kouyoumdjian JA, Morita Mda P, Araujo RG, et al. X-linked spinal and bulbar muscular atrophy (Kennedy's disease) with long-term electrophysiological evaluation: case report [J ]. Arq Neuropsiquiatr, 2005, 63(1) : 154-159.
[2]https://zhuanlan.zhihu.com/p/305781133
[3]俞立強,方琪,蔣覺安,許麗珍.肯尼迪病臨床、病理及遺傳學特點[J].臨床神經病學雜志,2015,28(04):296-298.
[4]Masahisa Katsuno,Hiroaki Adachi,Masahiro Waza,Haruhiko Banno,Keisuke Suzuki,Fumiaki Tanaka,Manabu Doyu,Gen Sobue. Pathogenesis, animal models and therapeutics in Spinal and bulbar muscular atrophy (SBMA)[J]. Experimental Neurology,2006,200(1).
[5]Constanza J. Cortes,Shuo-Chien Ling,Ling T. Guo,Gene Hung,Taiji Tsunemi,Linda Ly,Seiya Tokunaga,Edith Lopez,Bryce L. Sopher,C. Frank Bennett,G. Diane Shelton,Don W. Cleveland,Albert R. La Spada. Muscle Expression of Mutant Androgen Receptor Accounts for Systemic and Motor Neuron Disease Phenotypes in Spinal and Bulbar Muscular Atrophy[J]. Neuron,2014,82(2).
[6]Carlo Rinaldi,Laura C. Bott,Kenneth H. Fischbeck. Muscle Matters in Kennedy’s Disease[J]. Neuron,2014,82(2).
[7]https://www.nrdrs.org.cn/app/rare/disease-list-article.html?index=109
[8]馬俊芳,崔麗英,崔博.肯尼迪病的臨床特點、發病機制和治療進展[J].中華神經科雜志,2015,48(04):344-347.
| 上一篇:研究揭示PIWI亞家族蛋白在哺乳動物配子發生和早期胚胎發育中的非冗余功能 | 下一篇:猴痘病毒的感染與猴痘的動物模型 |
|
|
|
|
